
「薬事担当に配属されたけど、何から勉強すればいいか分からない」
「会議で『QMS』『GVP』が当たり前のように飛び交うけれど、ついていけない」
――そんな悩みを抱えていませんか?
本記事では、薬機法の骨格を実務目線に整理しました。
押さえるべきは、以下の3つの観点です。
- 規制対象(医療機器の定義・クラス分類)
- 市場投入の手続き(届出・認証・承認)
- 販売後の管理(QMS・GVP)
この3つを理解すれば、薬機法の全体像が理解できます。
1. 薬機法とは?正式名称と成り立ち

薬機法の正式名称は「医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律」です。
もともとは「薬事法(やくじほう)」という名称でしたが、2014年(平成26年)11月25日の改正施行によって現在の名称に変わりました。
「薬機法」という略称はこの改正後に使われるようになったもので、現在では業界内での標準的な呼び方になっています。
法律の目的は、医薬品・医療機器等の「品質」「有効性」「安全性」を確保すること、そして保健衛生上の危害の発生と拡大を防止することが主な目的です(第1条)。
2. 薬機法が規制する品目

薬機法は医療機器だけを対象とした法律ではありません。
大きく分けて以下の品目を規制しています。
| 品目 | 主な例 |
|---|---|
| 医薬品 | 処方薬、市販薬(OTC医薬品)など |
| 医薬部外品 | 薬用歯磨き、育毛剤、制汗剤など |
| 化粧品 | 日焼け止め、口紅、ファンデーションなど |
| 医療機器 | 診断・治療・予防に使用する機械器具等(本記事のメインテーマ) |
| 体外診断用医薬品 | 血糖測定試薬、PCR試薬、妊娠検査薬など |
| 再生医療等製品 | 細胞加工製品、遺伝子治療製品など |
※ 医療機器担当者が主に関わるのは「医療機器」と「体外診断用医薬品(体診)」です。
ただし法律全体が一つの体系として構成されているため、全体像を把握しておくことが重要です。
3. 医療機器の定義
薬機法における「医療機器」の定義は、第2条第4項に規定されています。
ARTICLE 2-4 — LEGAL DEFINITION
医療機器の定義(薬機法 第2条第4項)
人若しくは動物の疾病の診断、治療若しくは予防に使用されること、又は人若しくは動物の身体の構造若しくは機能に影響を及ぼすことが目的とされている機械器具等(再生医療等製品を除く。)であって、政令で定めるもの
ポイントは「目的」が規制の判断基準になっているという点です。
同じ機器でも、医療目的で使用されるかどうかによって薬機法の適用範囲が変わります。
たとえば、スマートウォッチの心拍センサーは一般的な健康管理目的であれば薬機法の対象外ですが、不整脈の診断を目的として使用する場合は医療機器として規制を受けます。
近年のデジタルヘルス領域では、この「目的」の判断が特に重要になっています。
なお、アプリやソフトウェア単体であっても、この定義に当てはまれば「プログラム医療機器(SaMD)」として医療機器に該当します。詳しくは「プログラム医療機器(SaMD)とは?薬機法の対象になる条件をゼロから解説」をご覧ください。
4. 医療機器のクラス分類(リスク別4区分)

薬機法では、医療機器をリスクの大きさに応じてクラスI〜IVの4つに分類しています。このクラスによって、製品の市場投入に必要な手続きが大きく異なります。
| クラス | 名称 | リスク | 主な例 | 必要な手続き |
|---|---|---|---|---|
| クラスI | 一般医療機器 | 極小 | X線フィルム、メス(柄) | 届出のみ |
| クラスII | 管理医療機器 | 小〜中 | MRI装置、電子血圧計 | 第三者認証機関による認証 (一部承認) |
| クラスIII | 高度管理医療機器 | 中〜高 | 透析器、人工呼吸器、コンタクトレンズ | PMDAを経た厚生労働大臣の承認 (一部認証) |
| クラスIV | 高度管理医療機器 | 高 | ペースメーカー、冠動脈ステント | PMDAを経た厚生労働大臣の承認 |
5. 製造・販売に必要な許可

医療機器を日本市場に流通させるためには、製品ごとの承認等だけでなく、事業者としての許可・登録も別途必要です。この2つをセットで取得しなければ販売できない点は、実務上の重要なポイントです。
① 製造販売業の許可
医療機器を「市場に出す(製造販売する)」事業者には、薬機法第23条の2に基づき、許可が必要です。
許可には以下の3種別があり、取り扱うクラスによって必要な種別が異なります。
| 許可種別 | 取り扱い可能なクラス | 主な要件 |
|---|---|---|
| 第一種医療機器製造販売業 | クラスI〜IV | QMS体制省令・GVP省令への適合 |
| 第二種医療機器製造販売業 | クラスI・II | QMS体制省令・GVP省令への適合 |
| 第三種医療機器製造販売業 | クラスIのみ | QMS体制省令への適合(要件簡略化)/GVP省令への適合 |
製造販売業者は製品の品質・安全性に最終的な責任を負う立場です。
そのため、製造販売業者は3つの責任者の設置が義務付けられています。
- 総括製造販売責任者(全体統括/薬機法施行規則 第114条の49)
- 国内品質業務運営責任者(品質管理/QMS体制省令 第3条)
- 医療機器安全管理責任者(市販後安全管理/GVP省令 第6条)
QMS SUPPLEMENT — QMS省令の管理層
QMS省令(製品の品質管理基準)の適用がある製造販売業者には、上記3責任者に加えて、QMSを構築・運用するための以下の役職も必要です。
- 管理監督者(Top Management):経営トップによるQMSの最終責任(QMS省令 第8条)
- 管理責任者(Management Representative):QMSの実務統括(管理監督者が任命/QMS省令 第19条)
- その他の管理者:設計開発・製造・品質管理の各責任者
※ ISO 13485 の管理層に対応する役職です。
② 製造業の登録
医療機器の製造(組立・包装・滅菌・設計等)を行う工場には、製造業の登録が必要です。
製造販売業の「許可」とは異なり、製造業は「登録」制度です。
また、海外の製造工場の場合は「外国製造業者の登録」が必要になります。
6. 承認・認証・届出の3つのルート
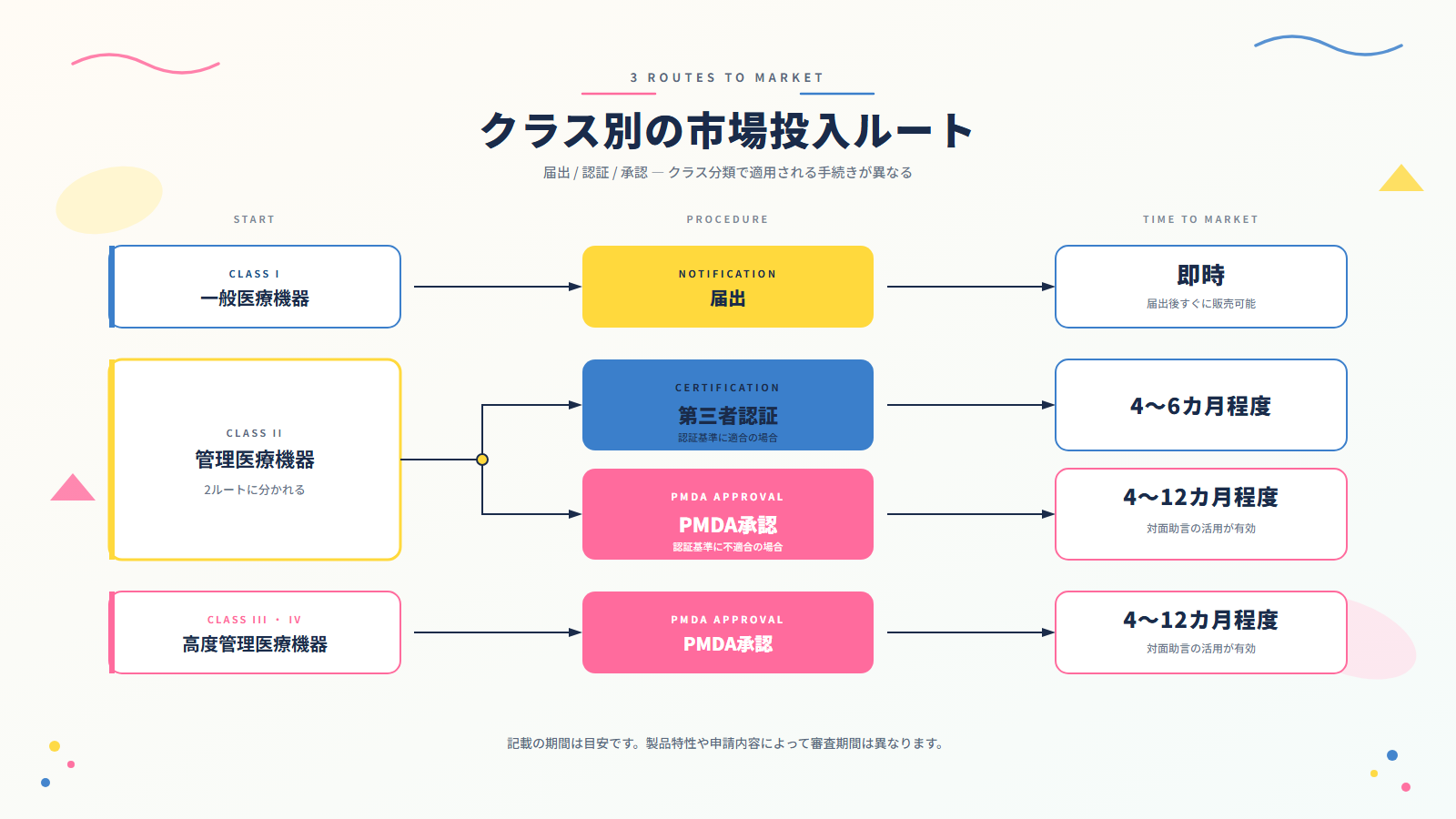
クラス分類に応じて、製品を市場に出すための手続きが異なります。
承認(クラスIII・IV、および一部のクラスII)
PMDA(独立行政法人 医薬品医療機器総合機構)が申請内容を審査し、厚生労働大臣が承認を与えます。
審査期間は製品の複雑さによって異なりますが、申請から承認まで通常4〜12カ月程度かかります。
事前に「対面助言(相談)」を活用することで、審査の効率化を図ることができます。
認証(クラスIIの多く)
厚生労働大臣が指定した第三者認証機関(登録認証機関)が審査します。
PMDAによる審査と比べて短期間で完了することが多く、申請から認証まで4〜6カ月程度が目安です。
ただし、認証基準(認証基準省令)に適合する製品に限られます。
届出(クラスI)
PMDAへの事前審査は不要で、製造販売届出書を提出することで販売できます。
手続き上は最も簡便ですが、届出内容は製造販売業者の自己責任です。
※ 記載の審査期間はあくまで目安です。製品の特性、申請内容、既承認品目との比較データの整備状況などによって、実際の期間は大きく異なる場合があります。
7. 市販後の主な義務
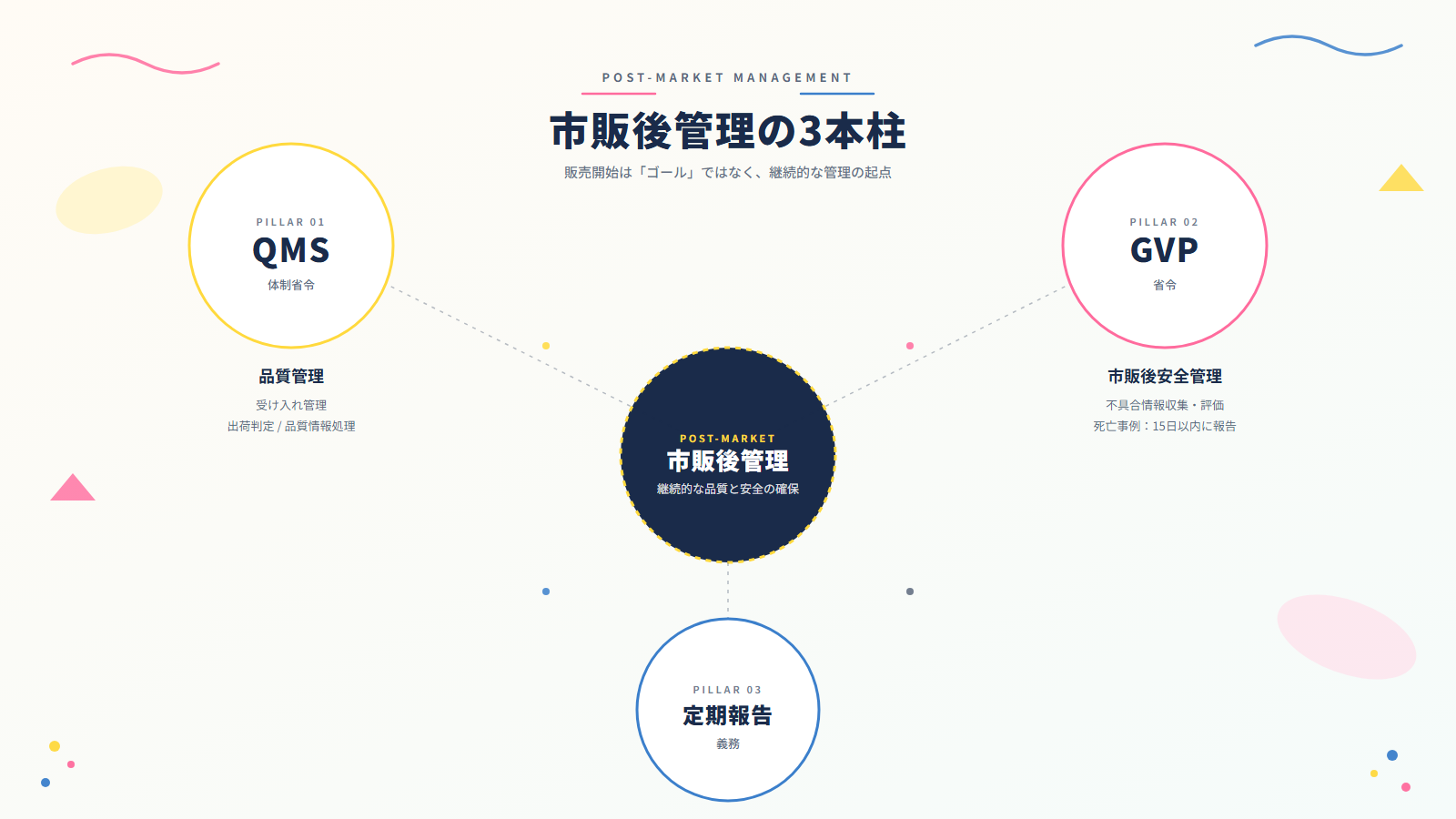
薬機法は承認・販売開始を「ゴール」とはしていません。
むしろ販売後も継続的な管理を求めており、この「市販後管理」こそが薬機法の重要な柱の一つです。
QMS体制省令(品質管理)
医療機器の製造販売業者が遵守すべき品質管理体制の基準を定めたものです(医療機器には GQP省令ではなく QMS体制省令・QMS省令 が適用されます)。
製造業者からの製品受け入れ管理・出荷判定・品質情報の収集・処理などが含まれます。
GVP省令(市販後安全管理)
市販後の不具合情報収集・評価・報告の体制を定めたものです。
不具合や感染症の報告期限が法定されており、たとえば死亡事例は15日以内、重篤な健康被害は30日以内の報告が義務付けられています。
定期報告
製造販売業者は、製品ごとに市販後の安全性情報を定期的にPMDAへ報告する義務があります(高度管理医療機器等は原則として毎年)。
まとめ:薬機法の骨格を3つの観点で整理
| 観点 | 内容 |
|---|---|
| ① 規制対象 | 医療機器はリスクに応じてクラスI〜IVに分類される。 クラスごとに必要な手続きが異なる。 |
| ② 市場投入の手続き | クラスに応じて「届出(クラスI)」「認証(クラスII)」「承認(クラスII〜IV)」の3ルート。 事業者は製造販売業の許可・製造業の登録も必要。 |
| ③ 販売後の管理 | QMS(品質管理)・GVP(市販後安全管理)・定期報告が継続的に義務付けられる。 死亡事例は15日以内など、報告期限が法定。 |
薬機法は法律本文だけで完結せず、その下にある省令(QMS・GVP・GCPなど)、告示、通知、ガイダンスが階層的に整備されています。
まずはこの記事で全体の骨格をつかんだうえで、自社の製品に関連する各省令・ガイダンスを深掘りしていくのが実務的なアプローチです。
参考法令・資料
- 医薬品、医療機器等の品質、有効性及び安全性の確保等に関する法律(昭和35年法律第145号)
- PMDA公式サイト:https://www.pmda.go.jp/
- 厚生労働省 医療機器ページ:https://www.mhlw.go.jp/stf/seisakunitsuite/bunya/kenkou_iryou/iryou/kiki/index.html


コメント