
医療機器を市場に出すには、製品ごとに 3 つの異なるルートが用意されています。
- 届出(製造販売届)
- 第三者認証機関の認証
- 厚生労働省の承認
「自社製品はどのルート?」
「認証と承認はどうやって確認するの?」
薬事担当者なら一度は迷う論点ではないでしょうか。
本記事では、薬機法の根拠条文から実務の判断ポイントまで、3 ルートの違いを解説します。
1. 医療機器の3つの申請ルートとは?

1-1. 承認・認証・届出の3区分とクラス分類の対応
医療機器を販売するためには、薬機法に基づき手続きが必要です。
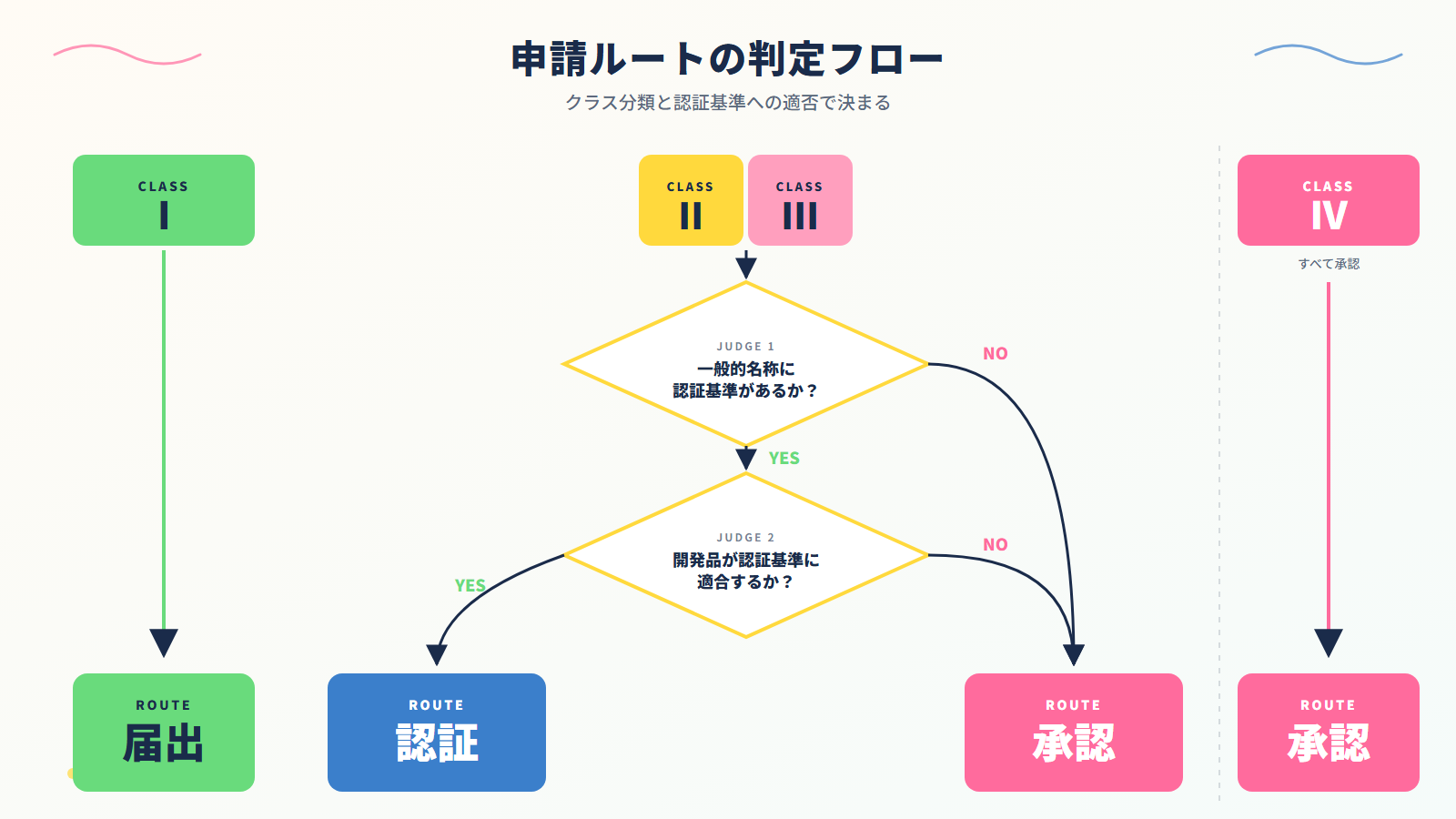
製品のクラス分類に応じて、申請ルートは届出・認証・承認の 3 つに分かれます。
クラス分類との対応関係は以下のとおりです。
| 申請ルート | 対応クラス | 申請先 |
|---|---|---|
| 届出 | クラスI(一般医療機器) | PMDA |
| 第三者認証 | クラスII・一部のクラスIII(認証基準あり) | 登録認証機関 |
| 厚生労働省承認 | クラスII〜IV(認証基準なし) | PMDAで審査 → 厚生労働大臣 |
クラスI は一律で届出、クラスIII の多くとクラスIV は 厚生労働省の承認が必要です。
判断が必要なのはクラスII とIIIで、認証基準の適否により2 ルートに分かれます。
1-2. なぜ3つに分かれているのか(薬機法の趣旨)
リスクと審査の効率性のバランスです。
すべてを PMDA 承認にすると審査が滞り、すべて届出にすると安全性が担保できません。
そこで、中間的リスクで認証基準により安全性が確保できる品目は民間の認証機関(登録認証機関)に審査を委託し、PMDA は新規性が高い品目や高リスク品の審査に集中する仕組みです。
この第三者認証制度は、2005年(平成17年)の改正薬事法(現・薬機法)で導入されました。
欧州の CE マーク制度とも類似する考え方を持つ制度です。
なお、対象がソフトウェア単体の場合は、申請ルートの前に「そもそも医療機器に該当するか(プログラム医療機器/SaMD)」の判断が必要です。詳しくは「プログラム医療機器(SaMD)とは?薬機法の対象になる条件をゼロから解説」をご覧ください。
2. 医療機器の届出(クラスI/一般医療機器)の実務
2-1. 届出とは?
届出は、薬機法で以下のとおり規定されています。
薬機法 第23条の2の12 第1項:
「製造販売業者は、一般医療機器の品目について、製造販売をしようとするときは、あらかじめ、その旨を厚生労働大臣に届け出なければならない。」
要するに「事前に届け出れば製造販売できる」最も簡易なルートです。
届出は、原則として承認審査のような技術的審査は行われず、主に記載内容や形式面の確認が実施されます。
2-2. 必要書類:製造販売届書の中身
製造販売届書に必要な記載事項は、以下のとおりです。
- 一般的名称(JMDN名称)
- 販売名
- 使用目的又は効果
- 形状、構造及び原理
- 原材料
- 性能及び安全性に関する規格
- 使用方法
- 保管方法及び有効期間
- 製造方法
- 製造販売する品目の製造所
詳細は、以下の通知を参照してください。
「医療機器の製造販売承認申請書添付資料の作成に際し留意すべき事項について」2015年(平成27年)1月20日 薬食機参発0120第9号(令和8年3月31日付け医薬機審発0331第7号により一部改正)
様式は、PMDAのホームページの「医療機器製造販売届」をダウンロードできます。
医療機器製造販売届の様式(PMDA)
2-3. 届出の流れと標準処理期間
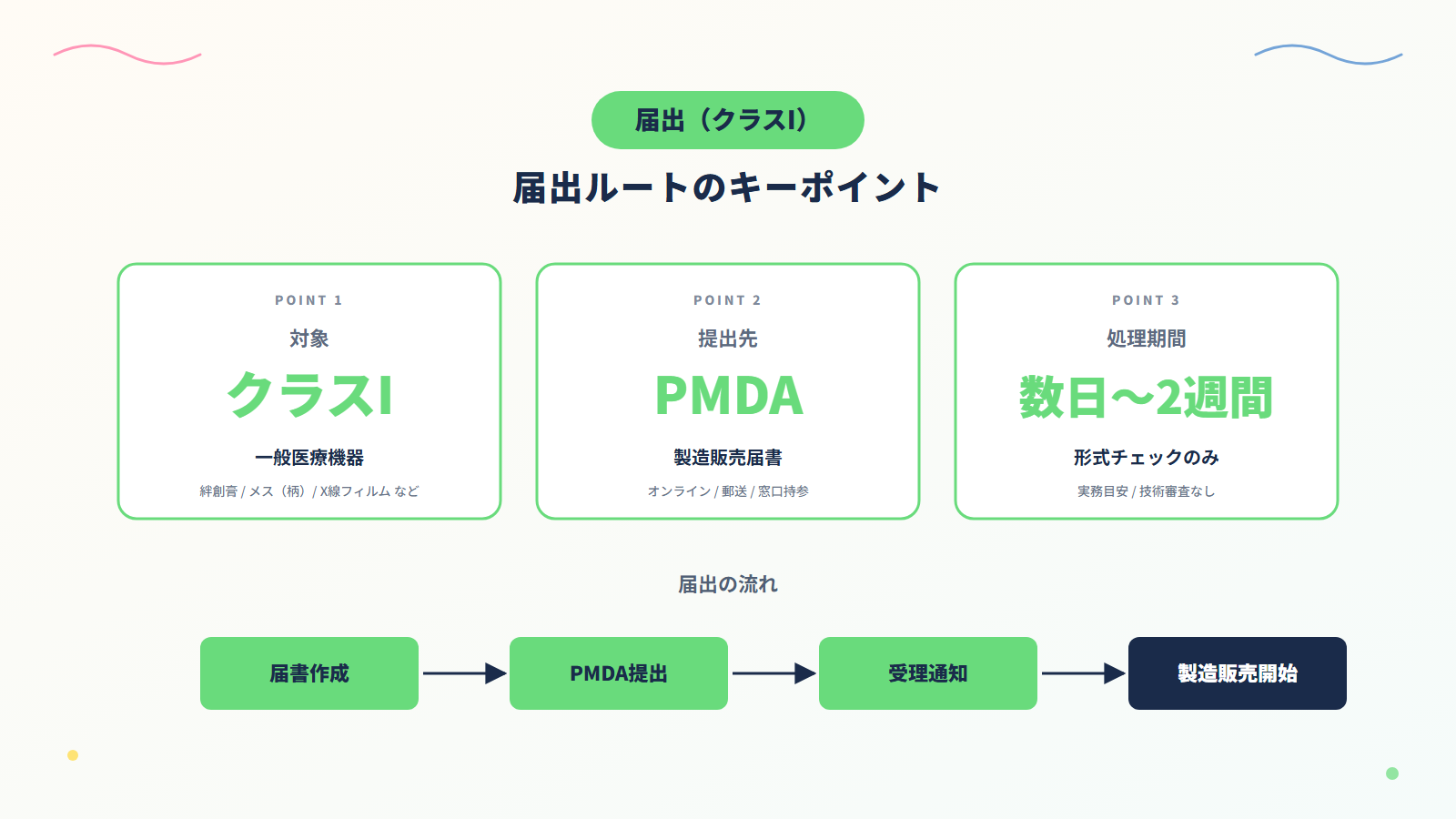
「医療機器製造販売届」を作成したら、PMDAに提出します。
提出方法は、以下の3つの方法があります。
- オンライン提出
- 郵送
- PMDAの受付窓口に持参
オンライン申請は、申請電子データシステム(ゲートウェイシステム)を使用します。
長期的に、複数の書類を提出する場合には、オンライン申請が印刷も不要で効率的ですが、システムの登録手続きや電子証明書の取得が必要になります。
オンライン申請について(PMDA)
あまり件数が多くない場合には、まずは郵送が簡単かもしれません。
届出書を提出後は、審査はなく、形式チェックのみです。
実務上、受理確認まで数日〜2週間程度(実務目安)です。
PMDAで受理されると、原則として製造販売が可能になります。
3. 医療機器の第三者認証(クラスII等/認証基準あり)の実務
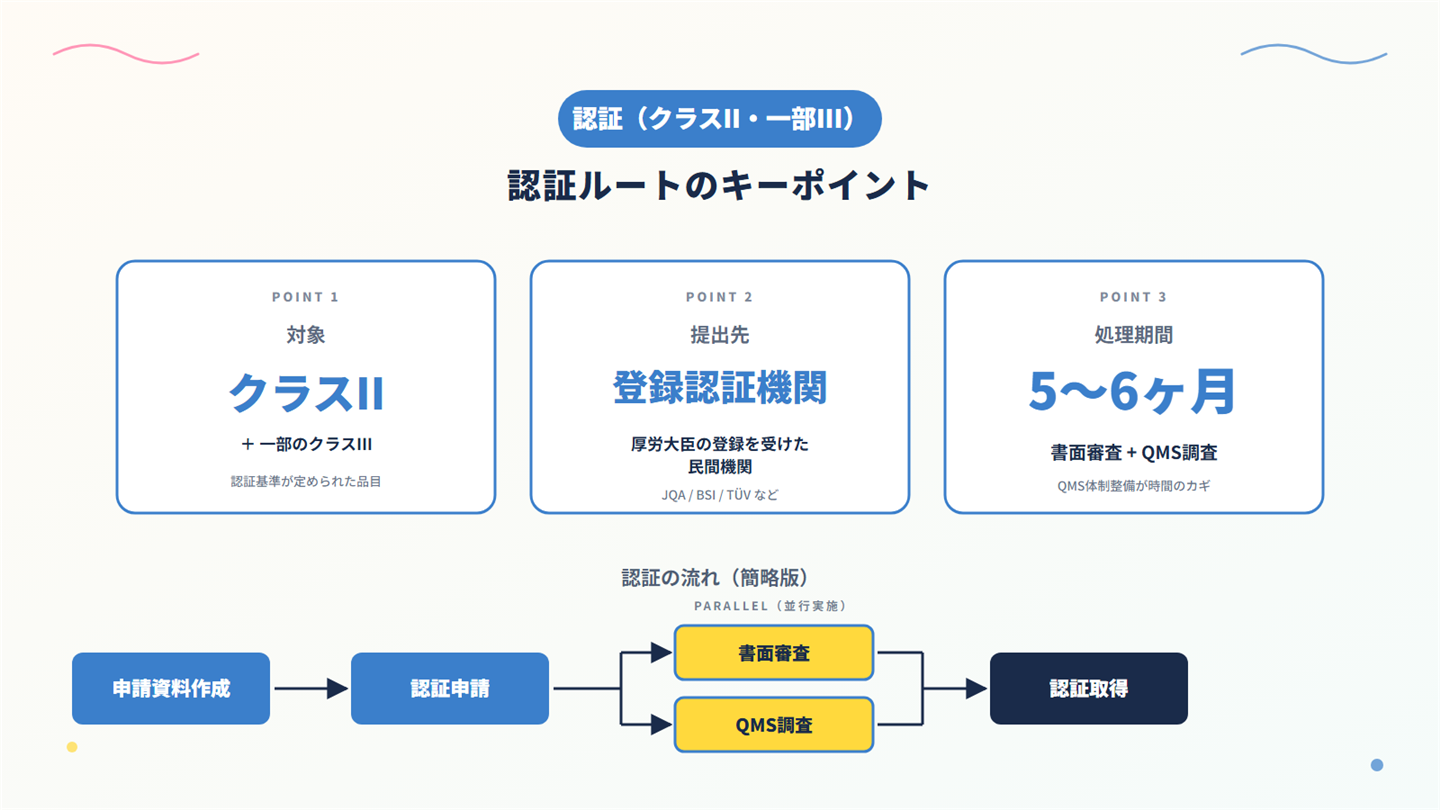
3-1. 登録認証機関の認証とは?
登録認証機関(第三者認証機関)の認証申請については、以下のとおり薬機法で規定されています。
薬機法 第23条の2の23(要約):
厚生労働大臣が基準を定めて指定する高度管理医療機器・管理医療機器(指定高度管理医療機器等)を製造販売するには、厚生労働大臣の登録を受けた登録認証機関の認証を受けなければならない、と規定されています。
つまり「認証基準が定められている品目」かつ「その基準に適合する製品」のみ、登録認証機関の認証で市場投入できる仕組みです。
3-2. 医療機器の認証基準とは何か
認証基準は、一般的名称(JMDN)ごとに厚生労働省告示で定められています。
内容は次のような要件で構成されます。
- 基本要件基準への適合(平成17年3月29日 厚生労働省告示第122号)
- JIS規格・IEC規格・ISO規格などの引用
- 安全性試験・性能試験の規格
- 使用目的・適応範囲の限定
代表例:自動電子血圧計の認証基準では、JIS T 1115(電子血圧計)、IEC 60601-1(電気医療機器の安全性)、IEC 60601-1-2(EMC)等への適合が求められます。
3-3. 登録認証機関の役割と選び方
認証基準に適合する場合には、登録認証機関に認証申請します。
登録認証機関は、厚生労働大臣の登録を受けた民間機関です。
登録認証機関は、厚生労働省のホームページで確認できます。
認証機関を選ぶ際のポイントは、以下のとおりです。
- 取り扱う製品の範囲(機関ごとに対応品目が異なる)
- 認証実績・経験(類似品目の認証経験)
- 標準処理期間
- 認証費用の見積もり
- QMS適合性調査の実施可能範囲(製造所が海外の場合は特に重要)
- 対応言語・サポート体制
取り扱う製品の範囲は、厚生労働省のホームページで確認できるので、最初に確認してください。
登録認証機関の認証範囲一覧(厚生労働省)
3-4. 認証申請の流れと標準処理期間
実務上の流れは、以下のとおりです。
- STEP 1:認証機関への事前相談(任意・推奨)
- STEP 2:申請資料の準備
- STEP 3:QMS適合性調査申請書の準備
- STEP 4:認証申請書・STED・QMS資料の提出
- STEP 5:認証機関による審査、QMS適合性調査
- STEP 6:認証取得
認証申請から認証取得まで の標準処理期間は、5〜6 ヶ月程度です(認証機関・品目により変動)。
QMS適合性調査の対応に時間がかかるケースが多いため、製造所側の体制整備を早めに進めることが重要です。
4. 医療機器の承認(クラスII〜IV/認証基準なし)の実務
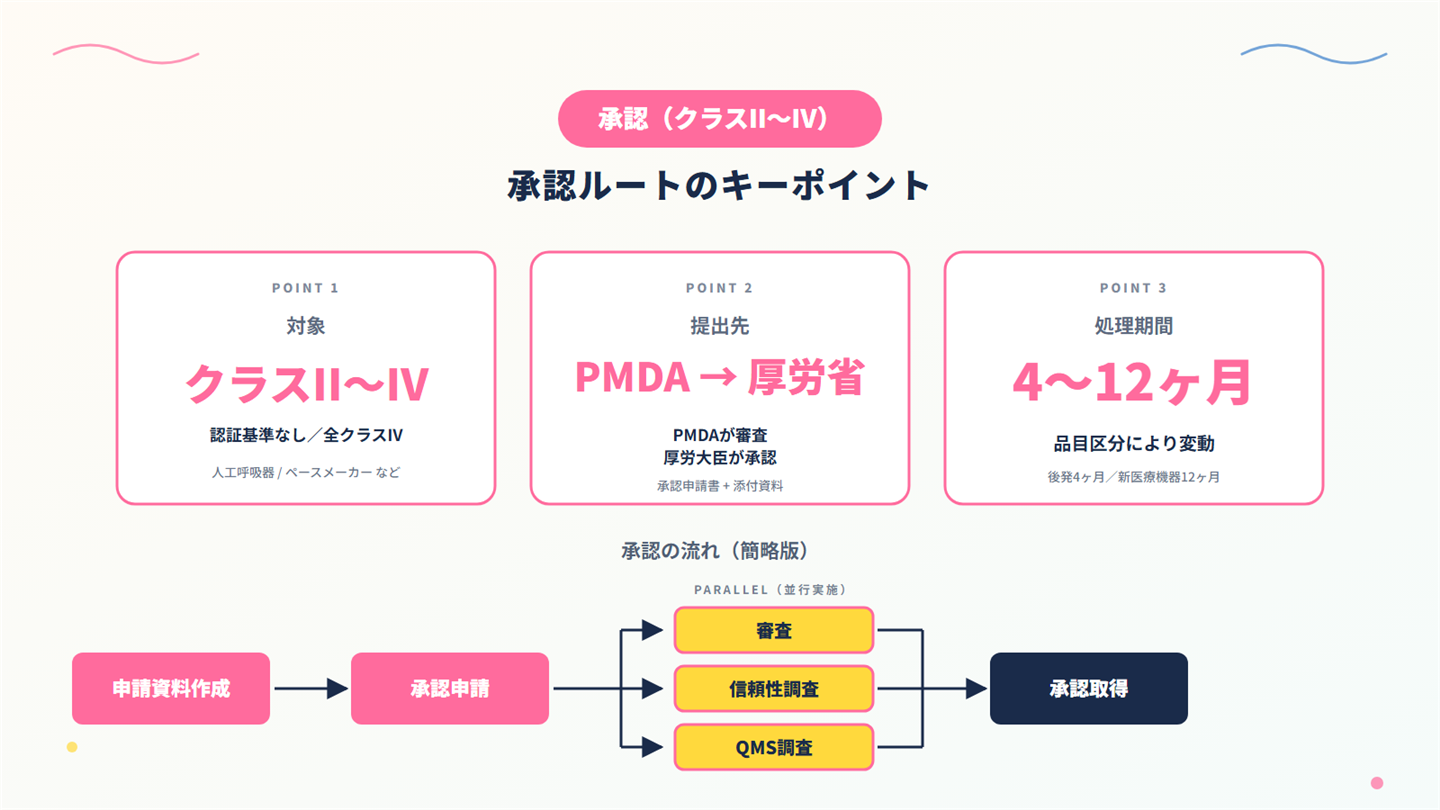
4-1. 厚生労働省の承認とは
厚生労働省の承認は、薬機法で以下のとおり規定されています。
薬機法 第23条の2の5 第1項:「医療機器(…略…)の製造販売をしようとする者は、品目ごとにその製造販売についての厚生労働大臣の承認を受けなければならない。」
実際の審査は PMDA(独立行政法人医薬品医療機器総合機構)が実施し、厚生労働大臣が承認を与えます。
クラスII とクラスIIIで認証基準に該当しない製品と、クラスIV が対象となります。
4-2. 承認申請に必要な資料
提出する資料は、医療機器の特性によって変わりますが、主な資料は以下のとおりです。
- 医療機器の申請書
- 添付資料
- 別添資料(試験報告書)
- 製造所に関する資料
提出方法は、「2-3. 届出の流れと標準処理期間」の届出方法と同じで、オンライン申請、郵送、PMDAの受付窓口のいずれかで提出します。
必要な書類は、以下の通知を参照してください。
4-3. 承認審査の流れと標準処理期間
承認申請の流れは、以下のとおりです。
申請前に必要な評価を確認するために、対面助言を利用することができます。
利用は任意ですが、利用することで審査が円滑に進む場合が多いです。
申請に必要な書類を準備します。
資料ができたら、PMDAに資料を提出します。
申請後は、PMDAによる審査、提出した試験の信頼性調査、製造所のQMS調査が行われます。
これらは、並行して行われることが多いです。
審査や調査で不明な点については、PMDAより照会事項が届くため、回答書で回答します。
内容にもよりますが1~2週間で回答を求められることが多いです。
新規性の高い品目で、医学専門家の確認が必要な場合には、専門協議が行われます。
また、新医療機器については、厚生労働省による部会が行われます。
実施はPMDAが行いますが、専門協議、部会のために資料の準備が求められます。
審査が終了すると、厚生労働省より承認がされます。
なお、標準処理期間は、以下のとおりです。
| 品目区分 | 標準処理期間 |
|---|---|
| 後発医療機器 | 4 ヶ月 |
| 改良医療機器(臨床試験なし) | 6 ヶ月 |
| 改良医療機器(臨床試験あり) | 9 ヶ月 |
| 新医療機器 | 12 ヶ月(優先審査は9カ月) |
5. 「認証 or 承認」の判定方法(クラスII の振り分け)
クラスII の製品では、認証基準があるかどうかで認証ルートと承認ルートが分かれます。判定の手順を整理します。
5-1. STEP 1:認証基準告示を確認する
対象品目の一般的名称(JMDN)に対して認証基準が設定されているかを確認します。
確認方法:開発品が該当する一般的名称に認証基準が設定されているかは、PMDAのホームページで確認することができます。
リンク:基準・JMDN検索データベース(PMDA)
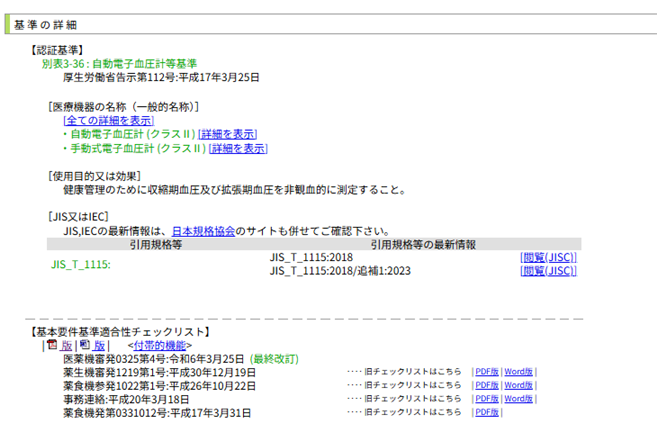
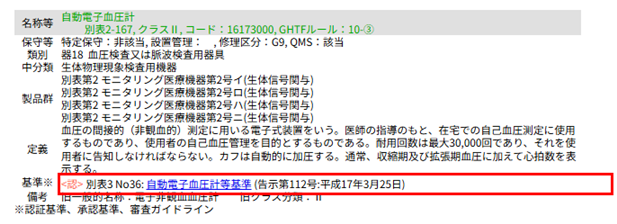
例えば、「自動電子血圧計」の場合、検索すると以下のように提示されます。
「基準※<認>別表〇〇~」とあれば、認証基準があります。
5-2. STEP 2:基準適合性を判断する
認証基準がある場合、自社製品が基準を満たすかを試験データ等で確認します。
確認すべき項目:
- 基本要件基準への適合
- 認証基準で引用される JIS / IEC / ISO 規格への適合
- 認証基準で定める使用目的・適応範囲の範囲内であること
- 認証基準の限定事項(例:単回使用、特定の構造に限る等)に該当しないこと
例えば、自動電子血圧計では、「別表3-36: 自動電子血圧計等基準」をクリックすると、適合する必要があるJIS規格などが分かります。
「基本要件基準適合性チェックリスト」を確認してください。
5-3. STEP 3:適合しない場合は承認ルートへ
次のようなケースでは認証ルートが使えず、PMDA 承認になります。
- 認証基準が設定されていない一般的名称
- 認証基準で限定された使用目的を逸脱する設計
- 新しい技術・材料を採用し、基準の規格に対応していない
判断に迷う場合は、まずは登録認証機関に相談します。
登録認証機関での判断が難しい場合には、PMDAの「認証基準該当性簡易相談」を活用できます。
ただし、PMDAに相談できるのは、「事前に登録認証機関に認証基準該当性を相談し、判断困難とされた品目」とされているため、一度は、登録認証機関に相談する必要があります。
6. 3ルート比較表で違いを一目で整理
届出・認証・承認の主な違いを表で整理します。
| 項目 | 届出 | 認証 | 承認 |
|---|---|---|---|
| 対応クラス | クラスI | クラスII・一部のクラスIII(認証基準に適合) | ・クラスII〜III(認証基準に非適合) ・クラスIV |
| 申請先 | PMDA | 登録認証機関 | PMDA→厚労省 |
| 業許可 | 第三種以上 | 第二種以上 | クラスIII・IV は第一種、クラスII は第二種以上 |
| 審査の有無 | 形式チェックのみ | 書面審査+QMS調査 | 書面審査+QMS調査+信頼性調査 |
| QMS適合性調査 | 簡略 | 必須 | 必須 |
| 標準処理期間 | 数日〜2週間程度(実務目安) | 5〜6 ヶ月 | 4〜12 ヶ月(品目区分による) |
なお、いずれのルートでも QMS体制の構築は必須です。
クラスI の届出でも QMS の整備義務はあり、市販後の品質管理・安全管理は同様に求められます。
7. よくある質問(FAQ)
まとめ:3ルートを「クラス × 認証基準」で判断する
医療機器の申請ルートは、クラス分類 × 認証基準の有無 で機械的に決まります。
- クラスI → 届出
- クラスIIとクラスIII(認証基準あり)→ 第三者認証
- クラスII・III・IV(認証基準なし) → PMDA 承認
3 ルートいずれにおいても、QMS体制の構築・基本要件基準への適合・リスクマネジメントは共通の前提です。違うのは「誰が審査するか」「どこまで詳しく審査するか」の部分です。
自社製品のルート判定で迷ったら、
- 認証基準がある場合は、登録認証機関
- 認証基準がない場合には、PMDA の全般相談
を早めに活用しましょう。
後から申請ルートが間違っていたと判明すると、開発スケジュールに大きな影響が出ます。
参考法令・資料
- 薬機法 第23条の2の5(承認)
- 薬機法 第23条の2の12(届出)
- 薬機法 第23条の2の23(認証)
- 薬機法施行規則 第114条の19以下(承認申請)
- 平成17年3月29日 厚生労働省告示第122号(基本要件基準)
- QMS省令(医療機器及び体外診断用医薬品の製造管理及び品質管理の基準に関する省令)
- PMDA「医療機器の標準処理期間」 https://www.pmda.go.jp/review-services/drug-reviews/about-reviews/devices/0035.html
- 厚生労働省告示 各認証基準告示


コメント